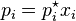Like distillation, operating lines can also be found in absorption. The basic principle to obtain the operating lines is the same. If we do the mass balance of the solute absorbed, we will end up with one equation, a linear equation.
Before we have a look at the mass balance, it is important to introduced a new term; loading. Loading is defined in the same way as molar fraction is. Normally a big capital X is used to represent loading, while a small one, x represents molar fraction.
Molar fraction, x = mole of solute / total mole available
Loading, X = mole of solute / (total mole available - mole of solute)
If molar fraction is given, it can simply be converted into loading via a very simple equation:
X = x/(1-x) or
x = X/(1+X)
Again X is for liquid phase and Y is for gas phase. There is however one important assumption to do the mass balance around the absorption column: Absorbent does not evaporate and carrier gas is not absorbed at the same time. This allows us to say that gas, G and liquid flow rate, L remain unchanged through out the column.

We know already G(in) = G(out) and it also valids L(in) = L(out)
Mass balance using loading:
G*Y(n+1) + L*Xo = G*Y1 + L*Xn
With some rearrangement we will get this final equation:
Y(n+1) = L/G *(Xn-Xo) + Y1
This is a straight line on a Y against X diagram.
Before we have a look at the mass balance, it is important to introduced a new term; loading. Loading is defined in the same way as molar fraction is. Normally a big capital X is used to represent loading, while a small one, x represents molar fraction.
Molar fraction, x = mole of solute / total mole available
Loading, X = mole of solute / (total mole available - mole of solute)
If molar fraction is given, it can simply be converted into loading via a very simple equation:
X = x/(1-x) or
x = X/(1+X)
Again X is for liquid phase and Y is for gas phase. There is however one important assumption to do the mass balance around the absorption column: Absorbent does not evaporate and carrier gas is not absorbed at the same time. This allows us to say that gas, G and liquid flow rate, L remain unchanged through out the column.

We know already G(in) = G(out) and it also valids L(in) = L(out)
Mass balance using loading:
G*Y(n+1) + L*Xo = G*Y1 + L*Xn
With some rearrangement we will get this final equation:
Y(n+1) = L/G *(Xn-Xo) + Y1
This is a straight line on a Y against X diagram.